HPC introduction
If you work at IBED you can get access to the Crunchomics HPC, the Genomics Compute Environment for SILS and IBED. If you need access to Crunchomics, send an email to Wim de Leeuw w.c.deleeuw@uva.nl to get an account set up by giving him your UvA netID.
Using an HPC works a bit differently than running jobs on your computer, below you find a simplified schematic:
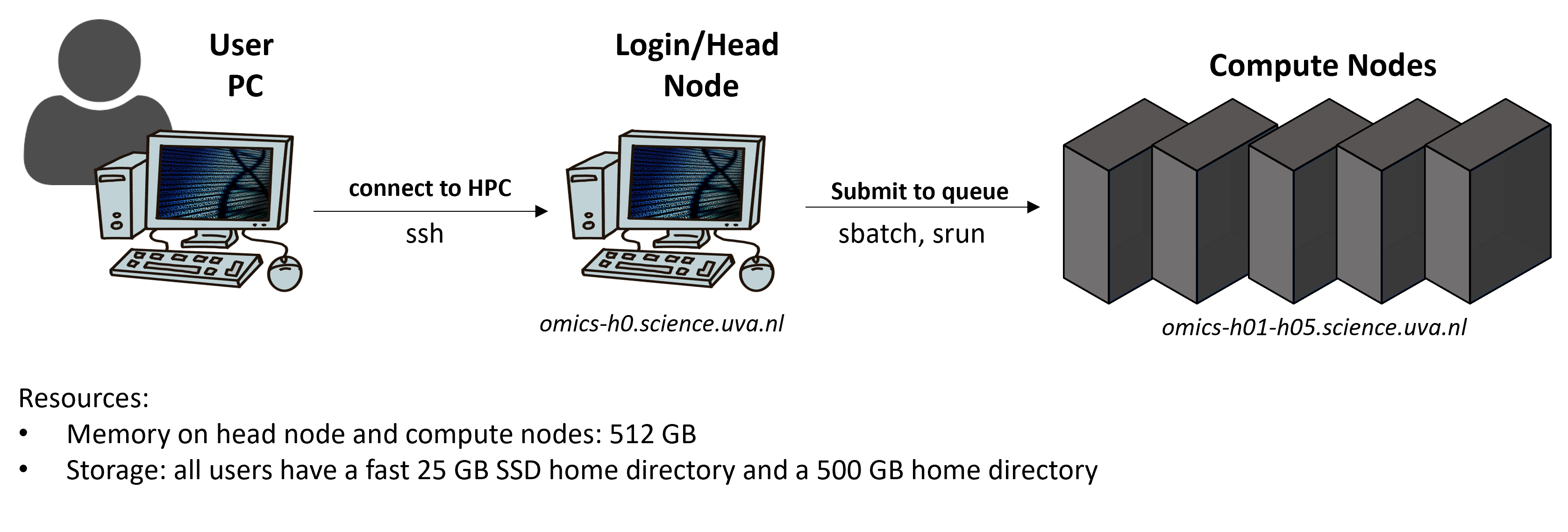
Very briefly, you can log into an HPC onto a login node. The purpose of a login node, sometimes also called head node, is to prepare to run a program (e.g., moving and editing files as well as compiling and preparing a job script). You then submit a job script from the head to the compute nodes via Slurm. The compute nodes are used to actually run a program and Slurm is an open-source workload manager/scheduler that is used on many big HPCs. Slurm has three key functions:
- provide the users access to the resources on the compute nodes for a certain amount of time to perform any computation
- provide a framework to start, execute, and check the work on the set of allocated compute nodes
- mange the queue of submitted jobs based on the availability of resources
Crunchomics etiquette
You share the HPC with other people, therefore, take care to only ask for the resources you actually use. Some general rules:
- There are no hard limits on resource usage, instead we expect you to keep in mind that you are sharing the system with other users. Some rules of thumb:
- Do NOT run jobs that request many CPUs and lots of memory on the head-node (omics-h0), use the compute nodes (omics-cn001 - omics-cn005) for this
- Do NOT allocate more than 20% (CPU or memory) of the cluster for more than a day. If you have large jobs, contact Wim de Leeuw
- Do not leave allocations unused and set reasonable time limits on you jobs
- For large compute jobs a job queuing system (SLURM) is available. Interactive usage is possible but is discouraged for larger jobs. We will learn how to use the queue during this tutorial
- Close applications when not in use, i.e. when running R interactively
On crunchomics you:
- Are granted a storage of 500 GB. After the duration of your grant, or when your UvAnetID expires, your data will be removed from the HPC. If you need more storage space, contact the Crunchomics team.
- In your home directory,
/home/$USER, you have 25 G quotum - In your personal directory,
/zfs/omics/personal/$USER, you can store up to 500 GB data - For larger, collaborative projects you can contact the Crunchomics team and ask for a shared folder to which several team members can have access
- You are in charge of backing up your own data and Crunchomics is NOT an archiving system. To learn about data archiving options at UvA visit the website of the computational support team
- A manual with more information and documentation about the cluster can be found here
Crunchomics gives you access to:
- 5 compute nodes
- Each compute node has 512 GB of memory and 64 CPUs
- If you need more resources (or access to GPUs), visit the website of the computational support team for more information
- Access to two directories:
- The home directory with 25 GB of storage
- your personal directory, with 500 GB of storage
Some information about storage:
- Snapshots are made daily at 00.00.00 and kept for 2 weeks. This means that if you accidentally remove a file it can be restored up to 2 weeks after removal. This also means that even if you remove files to make space, these files will still count towards your quota for two weeks
- Data on Crunchomics is stored on multiple disks. Therefore, there is protection against disk failure. However, the data is not replicated and you are responsible for backing up and/or archiving your data.
On the next page you find a tutorial to move the sequencing data that we worked on during the introduction into bash onto the server and run software to assess the quality of our sequencing data. We will learn to use some pre-installed software and also install software ourselves with conda and we will learn different ways to submit jobs to the compute nodes using SLURM.