An example notebook
If you want to see an example for documented code, check out a notebook I wrote based on what we are doing in this tutorial.
The link above leads you to an example for:
- How could I use github to make my code available to others
- How a “code book” could lool like. The example you see in the folder is provided as a Quarto markdown file here and for convenience the report was also rendered as a html to make it easier to read for potential collaborators. To view the HTML report, you need to download it first.
The example is based on the data you analyse throughout this tutorial, so the actual code might only make sense after you have finished the tutorial. Feel free to revisit this page after you have written some code yourself.
Please note that this is just an example to get you started and such a report might look different depending on your needs.
Below you find some general hints for what to put into different sections that you can see in the qmd file.
YAML headers

Setting up your working directory

In one of the first sections of my notebools I add everything that a user that wants to use my workflow has to change and I try to standardize the code below, i.e. I try to write it in a way so that it does not need to be changed. This ensures that another user could just run my code as is without having to edit anything other than this first section. Typically things to add are:
- From where to start the analyses, i.e. the location of the working directory or project folder
- Custom paths, to for example for databases that need to be downloaded from the web or repositories
- Custom variables: In the example above I store the link to the data in a variable called
download_link, I then use the variable in the code below to download the data. By doing it this way I have one location in the code another person needs to change the code when for example the path to the data changes. The code below stays the same since I use thedownload_linkvariable and not the actual path any more. When writing code try to think ahead and minimize the number of instances where things need to be changed if for example the location to your data changes.
I tend to NOT add the information on how I use scp to log into an HPC to keep my user name and login information private.
Sanity checks
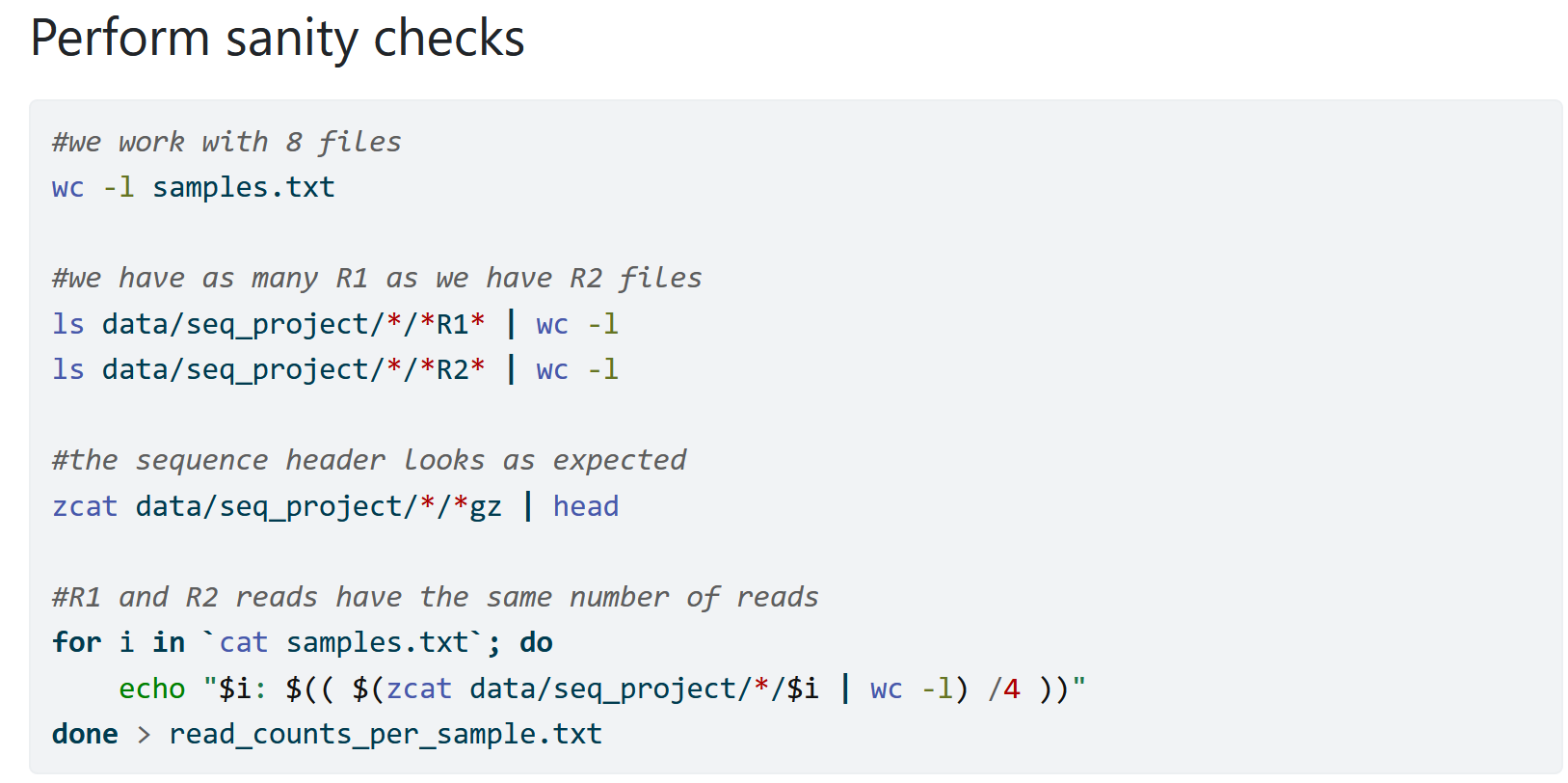
When I first start working with new data, I try to add as many sanity checks as possible to ensure that my data looks good. That way I avoid that I don’t notice an issue and run into trouble further down the line. I at the same time understand my data more and learn with how many samples and how much data I am working with.
I also add such sanity checks whenever I modify my data. For example, when I merge individual files into a large file I might count the lines for the individual files and the combined file simply to ensure that I used my wildcards correctly.
Remember: The computer is only as smart as the person using it and will blindly run your commands. Because of that the computer can do unexpected things and you need to account for that.
Using markdown to make notes

In the example above I use markdown to not only document my code but also add some comments whenever I might need it. In the example above, I added some notes about what the results from FastQC actually told me.
This kind of documentation can be useful for:
- Justifying decisions further down the line. In this example, I might decide on how to clean the sequencing data. For example, if I would have found a lot of low quality reads or the adapter still being part of the sequence then I would have specifically cleaned my sequences to deal with that
- Future you. If you read the report a month, or a year, later you have the key information in your report and don’t have to open any files or tables that are located elsewhere
Documenting external scripts
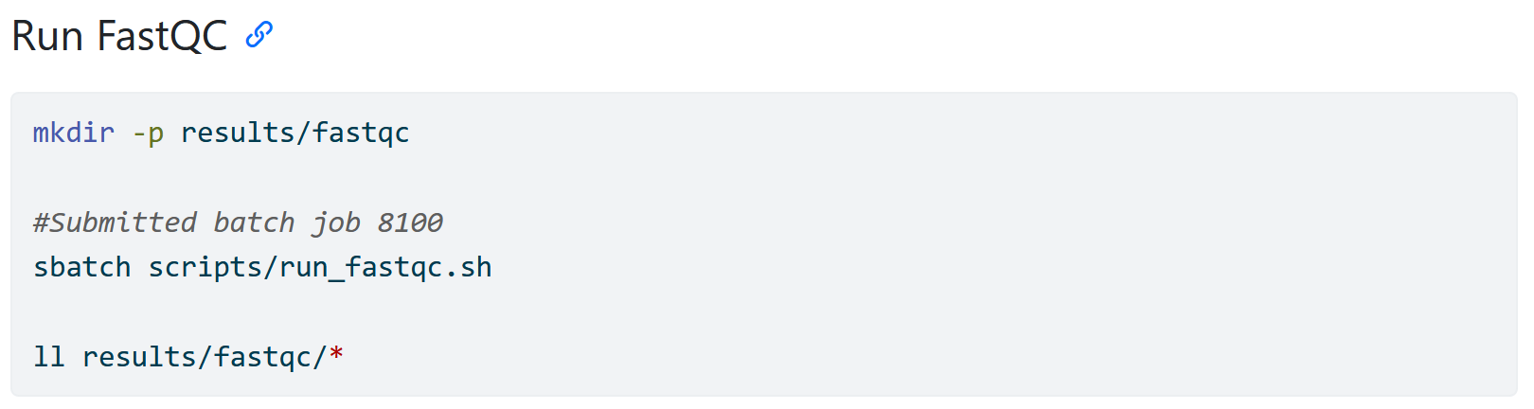
This part might make more sense after you have worked through the part of the tutorial about using an HPC. But what you see here is how I have written down code that simply says that I submitted a script to a HPC but it does not actually say how I ran the FastQC software. The actual code is “hidden” inside of the run_fastqc.sh script. This also means that a person reading your workflow does not have the code right away. You can deal with this in two ways.
- Instead of the sbatch command, you can add the actual line of code that was run on the compute node.
- When publishing your code with your manuscript, add the whole scripts folder to where you publish the main code, i.e. on github or zenodo
I tend to prefer number 2 because I like to record the code in my notebooks exactly as I ran it but you can do it differently as long as all the code you ran is recorded and accessible to others once you publish your data.