ssh -X username@serverUsing an HPC
Now, that you are familiar more familiar with the cli, let’s get used to working on an HPC by first login into Crunchomics, uploading our sequencing data an run some software to check the quality of our reads.
For people following this tutorial outside of UvA:
- If you have access to an HPC using SLURM you still follow the tutorial
- If you do not have access to an HPC but are interested in how to install and run software that can be used to analyse sequencing data, you still might want to check out the steps run below. The dataset is very small and all analyses can be run on a desktop computer
ssh: Connecting to a sever
SSH (Secure Shell) is a network protocol that enables secure remote connections between two systems. The general ssh command that you can use to login into any HPC looks as follows:
Options:
-Xoption enables untrusted X11 forwarding in SSH. Untrusted means = your local client sends a command to the remote machine and receives the graphical output. Put simply this option enables us to run graphical applications on a remote server and this for example allows us to view a pdf.
If you have access to and want to connect to Crunchomics you would edit the command above to look like this:
ssh -X uvanetid@omics-h0.science.uva.nl
Important
If you want to log into Crunchomics while working from UvA use the eduroam network, not the open Amsterdam Science Park network.
If you want to log into Crunchomics from outside of UvA you need to be connected to the VPN. If you have not set that up and/or have trouble doing so, please contact ICT.
Crunchomics: Preparing your account
Ignore this section if you already did this but if this is your first time on Crunchomics, then you want to first run a small Bash script that:
- Allows you to use system-wide installed software, by adding
/zfs/omics/software/binto your path. This basically means that bash knows that there is an extra folder in which to look for any software that is pre-installed on the HPC - Sets up a python3 environment and some useful python packages
- Have a link for your 500 GB personal directory in your home directory
To set this up, run the following command in the cli:
/zfs/omics/software/script/omics_install_scriptscp: Transferring data from/to a server
scp stands for Secure Copy Protocol and allows you to securely copy files and directories between remote hosts. When transferring data the transfer is always prepared from the terminal of your personal computer and not from the HPCs login node.
The basic syntax we use looks like this:
scp [options] SOURCE DESTINATION
To start analysing our data, we want to move the fastq.gz files that we have worked with before from our personal folder, the source, to a folder on Cruncomics, the destination. Let’s start setting up a project folder from which we want to run our analyses by:
- Moving from our home directory into our personal directory on the Crunchomics HPC. We move there since we have more space in the personal directory. Notice, for the command below to work, we need to have already executed the
omics_install_scriptscript above - Make a project folder with a descriptive file name, i.e. projectX
cd personal/
mkdir projectX
cd projectXNow that we have organized our working directory on the HPC, we next want to move the data folder (which right now is only on your own, personal computer) with the sequencing that you have downloaded before to Crunchomics. We do this by moving the whole data folder from our own computer to the new project folder on the HPC. Therefore, it is important that:
- we run the following command from the cli on our own computer and not from the cli while being logged into the HPC!
- you exchange the two instances of
usernamein the code below with your username/uvanetid - In the example below, I am running the code from inside of the
data_analysisfolder that we have generated in the previous tutorial since that is where I have the data folder that I want to move. I use the-roption in order to move the whole data folder and not a single file.
#run the command below from your local computer,
#ensure that the data folder is inside the directory from which you run this command
scp -r data username@omics-h0.science.uva.nl:/home/username/personal/projectX
#run ls on crunchomics to check if you moved the data successfully
ll data/seq_project/*/*fastq.gz
Tip: Moving data from the HPC to our own computer
We can also move data from the HPC to our own computer. For example, let’s assume we want to move a single sequencing file from crunchomics back to our computer. In this case,
- We do not need
-rsince we only move a single file - We again run this command from a terminal on our computer, not while being logged in the HPC
- We use
.to indicate that we want to move the file into the directory we are currently in. If we want to move the file elsewhere we can use any absolute or relative path as needed
scp username@omics-h0.science.uva.nl:/home/username/personal/projectX/data/seq_project/barcode001_User1_ITS_1_L001/Sample-DUMMY1_R1.fastq.gz .
Tip: Moving data from the HPC using wildcards
We can also move several files at once and ignore the whole folder structure by using wildcards when using scp.
#make a random directory in our Crunchomics working directory to move our data into
#again, we generate this folder on Crunchomics
mkdir transfer_test
#move files from crunchomics to our local computer
#again, always run scp from your local computer
scp data/seq_project/barcode00*/*fastq.gz username@omics-h0.science.uva.nl:/home/username/personal/projectX/transfer_test
#view the data, see how the folder structure is different compared to our first example?
#since we moved the data TO Crunchomics, we run ls while being logged into Crunchomics
ll transfer_test/*Notice for MAC users:
For Mac users that work with an zsh shell the command above might not work and you might get an error like “file not found”, “no matches found” or something the like. Without going into details zsh has a slightly different way of handling wildcards and tries to interpret the wildcard literally and thus does not find our files. If you see the error and you are sure the file exists it should work to edit your line of code as follows:
\scp data/seq_project/barcode00*/*fastq.gz username@omics-h0.science.uva.nl:/home/username/personal/projectX/transfer_testIf that does not work, these are some other things to try (different zsh environments might need slightly different solutions):
noglob scp data/seq_project/barcode00*/*fastq.gz ndombro@omics-h0.science.uva.nl:/home/ndombro/personal/projectX/transfer_test
scp 'data/seq_project/barcode00*/*fastq.gz' ndombro@omics-h0.science.uva.nl:/home/ndombro/personal/projectX/transfer_test
Tip: Moving files using the FileZilla GUI
scp takes some getting used to and there are several graphical user interface programs that are available which provide the same functionality. Here, we will look at FileZilla, as it is compatible with Windows, Mac, and Linux.
To use FileZilla, do the following:
- Download the FileZilla Client here. The free version is all we need.
- Follow the download instructions
- Click the FileZilla app to start it
- Add Uva user credentials
- Enter
omics-h0.science.uva.nlin host - Enter
user_namein user - Enter psswd chosen when making surf account
- Port should be 22 to open a sftp connection
- Enter
- Trust the host
- You land in your home directory (on the right hand side)
- You can copy-paste data by drag and dropping
Slurm basics
Get information about the cluster
Now, that we have our data prepared we want to run a tool to assess the quality of our reads. Before doing that, let’s talk about submitting jobs to an HPC.
As a reminder: We do not run big jobs on the login node but need to submit such jobs to the compute nodes via SLURM. Login nodes are for preparing your programs to run and you run your actual jobs by submitting them to the compute nodes using SLURM.
In the next few sections you will get to know the basics steps to be able to do this.
Before doing this it is, however, useful when getting started on a new HPC to know how to get basic information about what nodes are available on a cluster and how busy an HPC is. This allows us to better know how many resources to request in order to have our job run efficiently but also get started in a timely manner.
One way to get a list of available compute nodes is by typing the following command into the cli:
sinfoWe will see something like this:
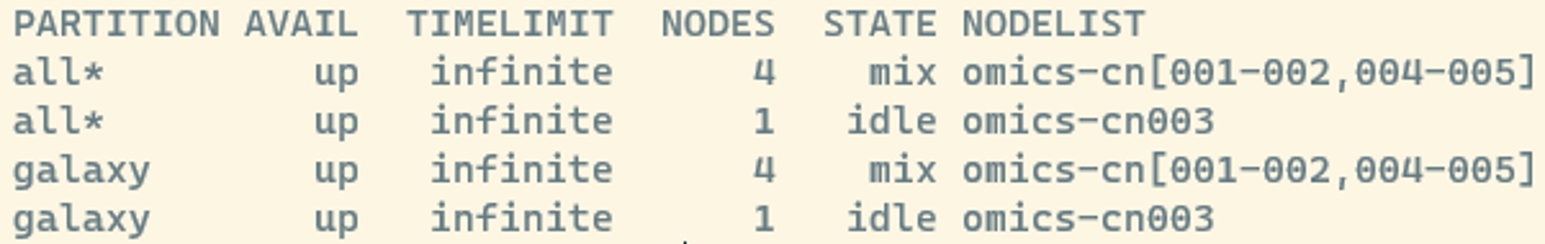
Here, the different columns you see are:
- partition: tells us what queues that are available. There are few partitions on Crunchomics and you do not need to define them in our case. However, other systems use partitions to group a subset of nodes with different type of hardwares or a specific maximum wall time. For example, you might have specific partitions for memory-heavy versus time-intensive jobs.
- state: tells you if a node is busy or not. Here:
- mix : consumable resources partially allocated
- idle : available to requests consumable resources
- drain : unavailable for use per system administrator request
- alloc : consumable resources fully allocated
- down : unavailable for use.
- Nodes: The number of nodes available for each partition
- NodeList: the names of the compute nodes (i.e. omics-cn001 to omics-cn005)
View info about jobs in the queue
The following command gives us some information about how busy the HPC is:
squeueAfter running this, you can see all jobs scheduled on the HPC, which might look something like this:
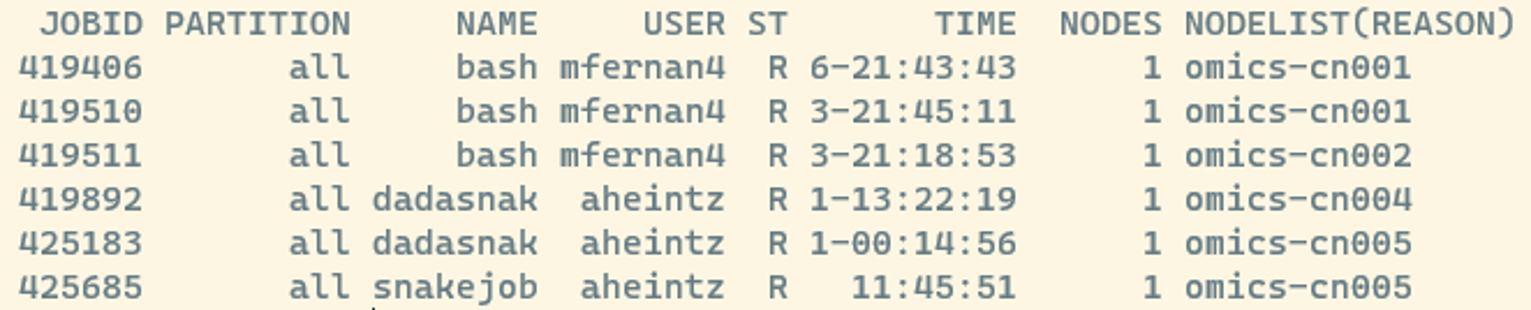
- JOBID: every job gets a number and you can manipulate jobs via this number
- ST: Job state codes that describe the current state of the job. The full list of abbreviations can be found here
If we would have submitted a job, we also should see the job listed there.
srun:Submitting a job interactively
srun is used when you want to run tasks interactively or want to have more control over the execution of a job. You directly issue srun commands in the terminal and you at the same time are able to specify the tasks to be executed and their resource requirements. For example, you might want to run softwareX and request that this job requires 10 CPUs and 5 GB of memory.
Use srun when:
- You want to run tasks interactively and need immediate feedback printed to the screen
- You are testing or debugging your commands before incorporating them into a script
- You need more control over the execution of tasks
- Typically, you use srun for smaller jobs that do not run for too long, i.e. a few hours
Let’s submit a very simple example for which we would not even need to submit a job, but just to get you started.
srun echo "Hello interactively"You should see the output of echo printed to the screen and if you would run squeue you won’t even see your job since everything ran so fast. But congrats, you communicated the first time with the compute node.
Now assume you want to run a more complex interactive task with srun that might run longer and benefit from using more CPUs. In this case you need to specify the resources your job needs by adding flags, i.e. some of which you see here:
srun --cpus-per-task=1 --mem=1G --time=00:10:00 echo "Hello interactively"The different flags mean the following:
--cpus-per-task=1: Specifies the number of CPUs per task. Adjust this based on the computational requirements of your task--mem=1G: Sets the memory requirement for the task. Modify this based on your task’s memory needs--time=00:10:00Sets the time limit to 10 minutesecho "Hello interactively: The actual command you want to run interactively
Other flags:
--nodes=1: Specifies the number of nodes. In this case, it’s set to 1 and tells slurm that we want to use a full node. Only use this if you make use of all resources on that node, otherwise omit. In our case this definitely is over-kill to request a full node with 64 CPUs to print a single line of text to the screen.--ntasks=1: Defines the number of tasks to run. Here, we would set it to 1 since we want to use echo once
Choosing the right amount of resources
When you’re just starting, deciding on the right resources to request for your computational job can be a bit challenging. The resource requirements can vary significantly based on the specific tool or workflow you are using. Here are some general guidelines to help you make informed choices:
- Use default settings: Begin by working with the default settings provided by the HPC cluster or recommended by the tool itself. These are often set to provide a balanced resource allocation for a wide range of tasks
- Check the software documentation: Consult the documentation of the software or tool you are using. Many tools provide recommendations for resource allocation based on the nature of the computation.
- Test with small datasets: For initial testing and debugging especially when working with large datasets, consider working with a smaller subset of your data. This allows for faster job turnaround times, helping you identify and resolve issues more efficiently.
- Monitor the resources usage:
- Use
sacctto check what resources a finished job has used and see whether you can optimize a run if you plan to run similar jobs over and over again. An example command would besacct -j 419847 --format=User,JobID,Jobname,state,start,end,elapsed,MaxRss,ncpus.In the report, look for columns like MaxRSS (maximum resident set size) to check if the amount of memory allocated (–mem) was appropriate. - Ensuer that the job used the resources you requested. For instance, if you would have used
--cpus-per-task=4 --mem=4G, you would expect to use a total of 16 GB of memory (4 CPUs * 4 GB). You can verify this withsacctto ensure your job’s resource requirements align with its actual usage.
- Use
- Fine-Tuning Resource Requests: If you encounter performance issues or your jobs are not completing successfully, consider iteratively adjusting resource requests. This might involve increasing or decreasing the number of CPUs, memory allocation, or other relevant parameters.
Run FastQC with srun
FastQC is a quality control tool for high throughput sequence data that is already installed on Crunchomics. This will be the actual software that we now want to run to look more closely at the quality of the sequencing data that we just uploaded to Crunchomics.
By the way: Whenever running a software for a first time, it is useful to check the manual, for example with fastqc -h.
Let’s start by setting up a clean folder structure to keep our files organized. I start with making a folder in which I want to store all results generated in this analysis and by using the -p argument I generate a fastqc folder inside the results folder at the same time. Then, we can submit our job using srun.
mkdir -p results/fastqc
srun --cpus-per-task=1 --mem=5G fastqc data/seq_project/*/*gz -o results/fastqc --threads 1
ll results/fastqc/*Since we work with little data this will run extremely fast despite only using 1 CPU (or thread, in our case these two words can be used interchangeably). However, if you would be logged into Crunchomics via a second window and run squeue you should see that your job is actively running (in the example below, the job we submitted is named after the software and got the jobid 746):
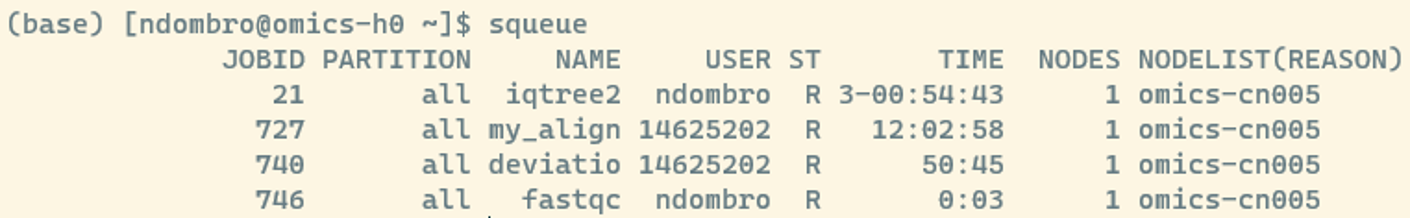
Additionally, after the run is completed, you should see that several HTML files were generated in our fastqc folder.
Exercise
Use scp to download the HTML files generated by fastqc to your own computer and view one of the HTML files.
Click me to see an answer
scp username@omics-h0.science.uva.nl:/home/username/personal/projectX/results/fastqc/*html results/fastqc/You could also open a file on Crunchomics with firefox results/fastqc/Sample-DUMMY1_R1_fastqc.html. However, connecting to the internet via a cli on a remote server tends to be rather slow and its often better to view files on your own computer especially if they are large files.
If you want to know more about how to to interpret the output, you can visit the fastqc website, which gives some examples for interpreting good and bad reports.
screen: Submitting long running jobs via srun
One down-side of srun for long running jobs is that your terminal gets “blocked” as long as the job is running and your job will be aborted if you loose the ssh connection to the HPC. For long running jobs, there are two ways to deal with this:
- submit a srun job in a screen
- use sbatch
In this section, we will cover how to use screen. Screen or GNU Screen is a terminal multiplexer. This means that you can start a screen session and then open any number of windows (virtual terminals) inside that session.
Processes running in Screen will continue to run when their window is not visible even if you get disconnected. This is perfect, if we start longer running processes on the server and want to shut down our computer when leaving for the day. As long as the server is still connected to the internet, your process will continue running.
We start a screen as follows:
screenWe detach (go outside of a screen but keep the screen running in the background) from a screen by pressing control+a+d.
If you want to run multiple analyses in multiple screens at the same time, then can be useful to give your screens more descriptive names. You can give screens a name using the -S option:
screen -S run_fastqcAfter detaching from this screen again with control+a+d you can create a list of all currently running screens with:
screen -lsYou can re-connect to an existing screen like this:
screen -r run_fastqcNow inside our screen, we can run fastqc same as we did before (but now don’t risk to loose a long-running job):
srun --cpus-per-task=1 --mem=5G fastqc data/seq_project/*/*gz -o results/fastqc --threads 1 For long-running jobs we can start multiple screens at once or even if we just have one screen open, close it and leave for the day or simply work on other things on the cli outside of the screen.
If you want to completely close and remove a screen, type the following and press enter while being inside of the screen:
exitsbatch: submitting a long-running job
sbatch is your go-to command when you have a script (i.e. a batch script) that needs to be executed without direct user interaction.
Use sbatch when:
- You have long-running or resource-intensive tasks
- You want to submit jobs that can run independently without your immediate supervision
- You want to submit multiple jobs at once
To run a job script, you:
- create a script that contains all the commands and configurations needed for your job
- use sbatch to submit this script to the Slurm scheduler, and it takes care of the rest.
Let’s start with generating some new folders to keep our project folder organized:
mkdir scripts
mkdir logsTo get started, assume we have created a script in the scripts folder named run_fastqc.sh with the content that is shown below. Notice, how in this script I added some additional echo commands? I just use these to print some information about the progress which could be printed to a log file but if you have several commands that should be executed after each other that is how you would do it.
#!/bin/bash
#SBATCH --cpus-per-task=1
#SBATCH --mem=5G
#SBATCH --time=1:00:00
echo "Start fastqc"
fastqc data/seq_project/*/*gz -o results/fastqc --threads 1
echo "fastqc finished"In the job script above:
#!/bin/bash. This so-called Shebang line tells the shell to interpret and run the Slurm script using the bash shell. This line should always be added at the very top of your SBATCH/Slurm script.- The lines that follow and start with
#are the lines in which we define the amount of resources required for our job to run. In our case, we request 1 CPU, 5G of memory and 1 hour time limit. - If your code needs any dependencies, such as conda environments, you would add these dependencies here. We do not need this for our example here, but you might need to add something like this
conda activate my_envif you have installed your own software. We will touch upon conda environments and installing software a bit later. - The lines afterwards are the actually commands that we want to run on the compute nodes, We also call this the job steps.
To prepare the script and run it:
- Run
nano scripts/run_fastqc.shto generate an empty jobscript file - Add the code from above into the file we just opened
- Press
ctrl+xto exit nano - Type
Ywhen prompted if the changes should be saved - Confirm that the file name is good by pressing enter
Afterwards, you can submit run_fastqc.sh as follows:
#submit job: 754
sbatch scripts/run_fastqc.sh
#check if job is running correctly
squeueAfter running this, you should see that the job was submitted and something like this printed to the screen Submitted batch job 754. You will also see that a new file is generated that will look something like this slurm-754.out.
When you submit a batch job using sbatch, Slurm redirects the standard output and standard error messages, which you have seen printed to the screen when you used srun, to a file named in the format slurm-JOBID.out, where JOBID is the unique identifier assigned to your job.
This file is useful as it:
- Captures the output of our batch scripts and stores them in a file
- Can be used for debugging, since if something goes wrong with your job, examining the contents of this file can provide valuable insights into the issue. Error messages, warnings, or unexpected outputs are often recorded here
Feel free to explore the content of the log file, do you see how the echo commands are used as well?
Tip: sbatch and better log files
We have seen that by default sbatch redirects the standard output and error to our working directory and that it decides itself how to name the files. Since file organization is very important especially if you generate lots of files, you find below an example to:
- Store the standard output and error in two separate files
- Redirect the output into another folder, the logs folder
- In the code below, the
%jis replaced with the job allocation number once the log files are generated
#!/bin/bash
#SBATCH --job-name=our_fastqc_job
#SBATCH --output=logs/fastqc_%j.out
#SBATCH --error=logs/fastqc_%j.err
#SBATCH --cpus-per-task=1
#SBATCH --mem=5G
#SBATCH --time=1:00:00
echo "Start fastqc"
fastqc data/seq_project/*/*gz -o results/fastqc --threads 1
echo "fastqc finished"
Advanced tip: sbatch arrays to run multiple files in parallel
With fastqc we are very lucky that the tool can identify all the fastq files in the directory we specify with -o by making use of the wildcard. This is extremely useful for us but by far not all programs work this way.
For this section, lets assume that we need to provide each individual file we want to analyse, one by one, and can not provide a folder name. How would we run such a job effectively?
What we want to do is created what is called a job array that allows us to:
- Run multiple jobs that have the same job definition, i.e. cpus, memory and software used
- Run these job in the most optimal way. I.e. we do not want to run one job after each other but we also want to run jobs in parallel at the same time to optimize resource usage.
Let’s start with making a list with files we want to work with based on what we have already learned:
ls data/seq_project/*/*.gz | cut -f4 -d "/" > samples.txtNext, we can use this text file in our job array, the content of which we store in scripts/array.sh:
#!/bin/bash
#SBATCH --job-name=my_array
#SBATCH --output=logs/array_%A_%a.out
#SBATCH --error=logs/array_%A_%a.err
#SBATCH --array=1-8
#SBATCH --cpus-per-task=1
#SBATCH --mem-per-cpu=5G
#calculate the index of the current job within the batch
#in our case the index will store the values 1 to 8 for our 8 files
INDEX=$((SLURM_ARRAY_TASK_ID ))
#build an array structure that stores the fastq.gz file names
CURRENT_SAMPLE=$(cat samples.txt | sed -n "${INDEX}p")
#print what is actually happening
echo "Now Job${INDEX} runs on ${CURRENT_SAMPLE}"
#run the actual job
fastqc data/seq_project/*/${CURRENT_SAMPLE} -o results/fastqc --threads 1In the script we use some new SLURM arguments:
#SBATCH --array=1-8: Sets up a job array, specifying the range (1-8). We choose 1-8 because we have exactly 8 fastq.gz files we want to analyse#SBATCH --output=logs/array_%A_%a.out: Store the standard output and error.%Arepresents the job ID assigned by Slurm, and%arepresents the array task ID- Important: Are your individual arrays large jobs that use a lot of CPUs/memory? Then you might not want to submit all at the same time but submit them in a step-wise manner. With
#SBATCH --array=1-8%2you would only submit the first two jobs and only after these have finished the next jobs. Remember, try to use not more than ~20% of the crunchomics cluster at the same time.
The job does the following:
- The
INDEXvariable is storing the value of the currentSLURM_ARRAY_TASK_ID. This represents the ID of the current job within the array. In our case this will be first 1, then 2, …, and finally 8. - Next, we build the array structure in which the
CURRENT_SAMPLEvariable is created by:- Reading the
sample_list.txtfile withcat - Using a pipe to extract the file name at the calculated index using sed. Sed is an extremely powerful way to edit text that we have not covered here but
-n 1pis a option that allows us to print one specific line of a file, in our case the first one when running array 1. So for the first array the actual code run is the followingcat samples.txt | sed -n "1p". For the next array, we would runcat samples.txt | sed -n "2p"and so forth. - The output of the pipe is stored in a variable, called
CURRENT_SAMPLE. For our first sample this will beSample-DUMMY1_R1.fastq.gz
- Reading the
- We use echo to record what was executed when and store this information in the standard output
- We run our actual fastqc job on the file name that is currently stored in the
CURRENT_SAMPLEvariable.
If we check what is happening right after submitting the job with squeue we should see something like this:
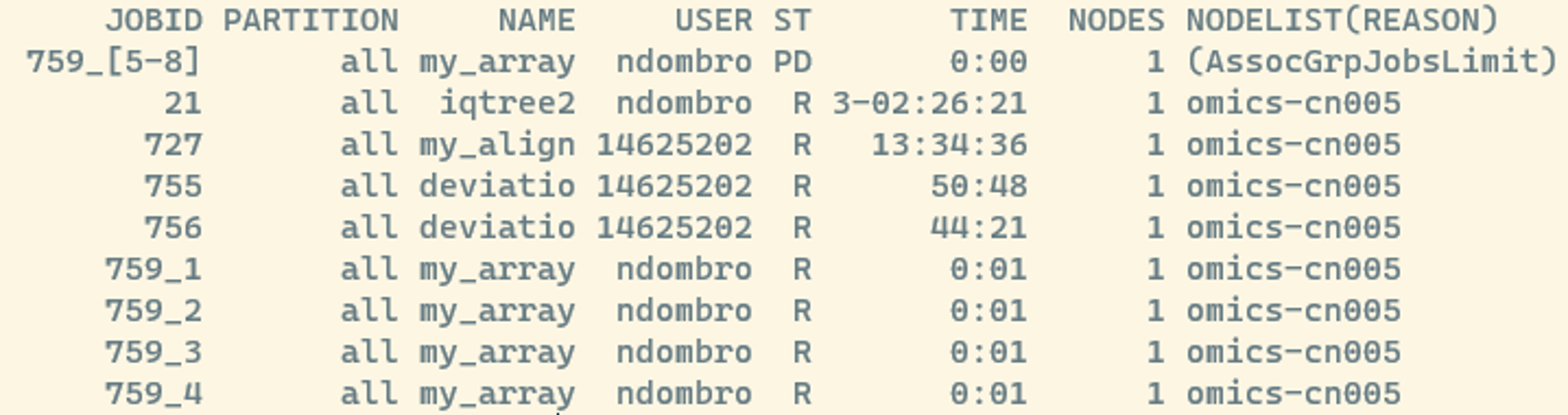
We see that jobs 1-4 are already running and the jobs 5-8 are currently waiting for space. That is one of the useful things using a job manager such as SLURM. It takes care of finding the appropriate resources on all nodes for us as long as we defined the required cpus and memory sensibly.
If we check the log files we should see something like this:
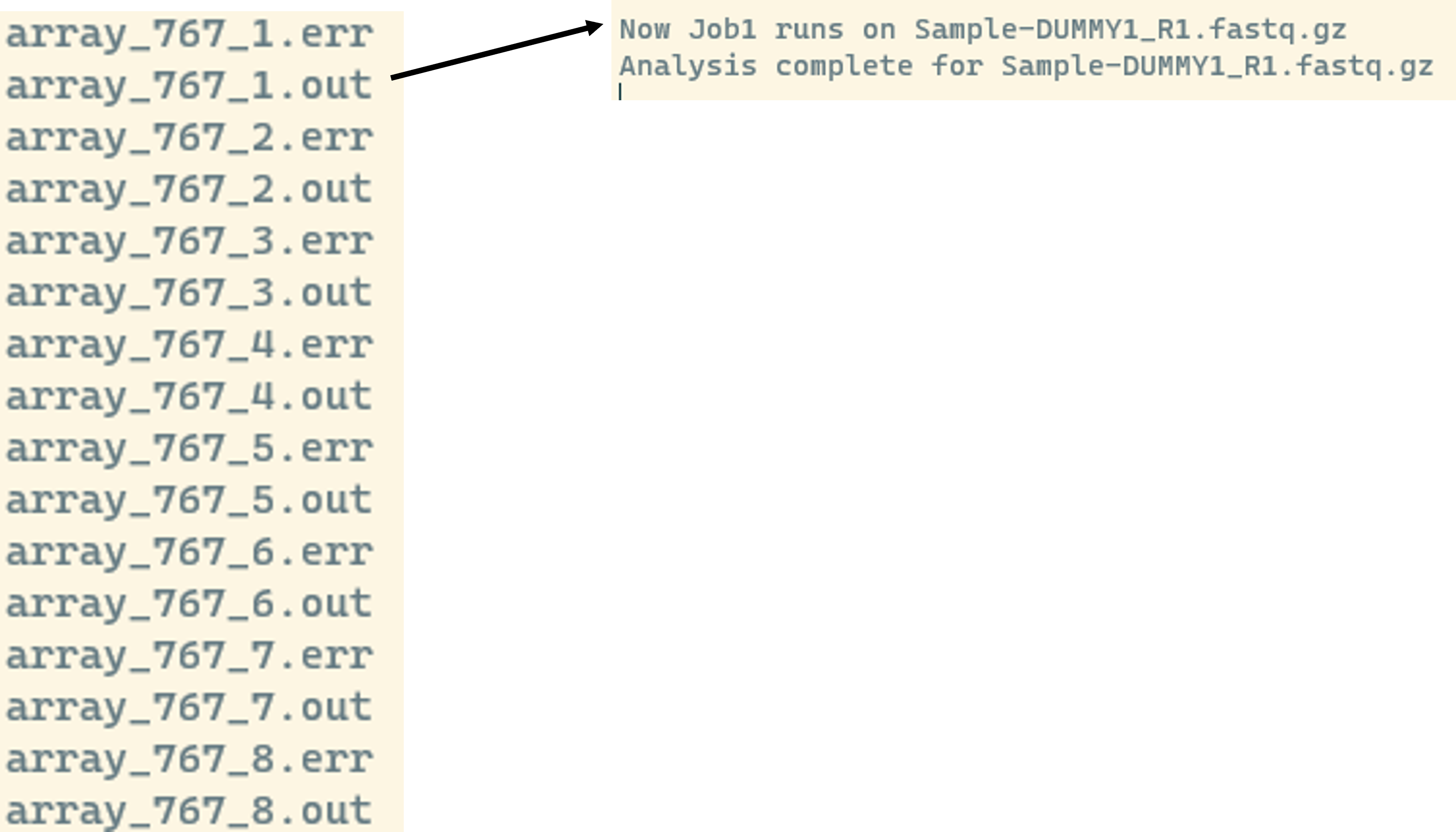
We see that we get an individual output and error file for each job. In the output we see what value is stored in the INDEX, here 1, and the CURRENT_SAMPLE, here Sample-DUMMY1_R1.fastq.gz and that the analysis finished successfully.
Installing software
There might be cases where the software you are interested in is not installed on the HPC you are working with (or on your own computer).
In the majority of cases, you should be able to install software by using a package management system, such as conda or mamba. These systems allow you to find and install packages in their own environment without administrator privileges. Let’s have a look at a very brief example:
Install conda/mamba
A lot of systems already come with conda/mamba installed, however, if possible we recommend working with mamba instead of conda. mamba is a replacement and uses the same commands and configuration options as conda, however, it tends to be much faster. A useful thing is that if you find documentation for conda then you can swap almost all commands between conda & mamba.
If you have conda installed and do not want to install anything else, that is fine. Just replace all instances of mamba with conda below.
This command should work in most cases to setup conda together with mamba:
curl -L -O "https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-$(uname)-$(uname -m).sh"
bash Miniforge3-$(uname)-$(uname -m).shWhen running the bash command, you get asked a few questions:
- Read the license and use arrow down to scroll down. Don’t scroll too fast, so that you see the next question
- Decide where conda gets installed. You can say yes to the default location in your home. But don’t forget that for Crunchomics your home only has 25G of space. You could also install the miniforge/mambaforge folder in your personal folder instead.
- Say yes, when you get asked whether conda should be initialized during start up
- Restart the shell (exit the terminal and use ssh to log back in) for the changes to take effect
- Check if conda is accessible by running
conda -h
Setting up an environment
Let’s assume we want to install seqkit, a tool that allows us to calculate some statistics for sequencing data such as the number of sequences or average sequence length per sample.
We can install seqkit into a separate environment, which we can give a descriptive name, as follows:
#check if the tool is installed (should return "command not" found if the software is not installed)
seqkit -h
#create an empty environment and name it seqkit and we add the version number to the name
#this basically sets up seqkit separate from our default working environment
#this is useful whenever software require complicated dependencies allowing us to have a separate install away from software that could conflict with each other
mamba create -n seqkit_2.6.1
#install seqkit, into the environment we just created
mamba install -n seqkit_2.6.1 -c bioconda seqkit=2.6.1
#to run seqkit, we need activate the environment first
mamba activate seqkit_2.6.1
#check if tool is installed,
#if installed properly this should return some detailed information on how to run seqkit
seqkit -h
#run the tool via srun
mkdir results/seqkit
srun --cpus-per-task 2 --mem=4G seqkit stats -a -To results/seqkit/seqkit_stats.tsv data/seq_project/*/*.gz --threads 2
less -S results/seqkit/seqkit_stats.tsv
#close the environment
conda deactivateWhen installing seqkit, we:
- specify the exact version we want to download with
=2.6.1. We could also install the newest version that conda/mamba can find by runningmamba install -n seqkit -c bioconda seqkit. - specify that we want to look for seqkit in the bioconda channel with the option
-c. Channels are the locations where packages are stored. They serve as the base for hosting and managing packages. Conda packages are downloaded from remote channels, which are URLs to directories containing conda packages. If you are unable to find a package it might be that you need to specify a channel.
Forgot what conda environments you installed in the past? You can run conda env list to generate a list of all existing environments.
Unsure if a software can be installed with conda? Google conda together with the software name, which should lead you do a conda web page. This page should inform you whether you need to add a specific channel to install the software as well as the version numbers available.
A full set of mamba/conda commands can be found here.
Exercise
- Download and view the file
results/seqkit/seqkit_stats.tsvon your own computer - Run the seqkit again but this time submit the job via a sbatch script instead of using srun. Notice, that you need to tell SLURM how it can activate the conda environment that has seqkit installed. You might need to google how to do that, since this requires some extra line of code that we have not covered yet but see this as a good exercise for how to handle error messages that you see in the log files for your own analyses
Click me to see an answer
#question 1
scp username@omics-h0.science.uva.nl:/home/ndombro/personal/projectX/results/seqkit/seqkit_stats.tsv .
#question 2
sbatch scripts/seqkit.shContent of scripts/seqkit.sh:
#!/bin/bash
#SBATCH --job-name=seqkit_job
#SBATCH --output=logs/seqkit_%j.out
#SBATCH --error=logs/seqkit_%j.err
#SBATCH --cpus-per-task=2
#SBATCH --mem=5G
#activate dependencies
source ~/.bashrc
mamba activate seqkit_2.6.1
#run seqkit
echo "Start seqkit"
seqkit stats -a -To results/seqkit/seqkit_stats.tsv data/seq_project/*/*.gz --threads 2
echo "seqkit finished"In the script above, we see that we need to add two lines of code to activate the seqkit conda environment:
source ~/.bashrc
mamba activate seqkit_2.6.1When you run a script or a command, it operates in its own environment. The source command is like telling the script to look into another file, in this case, ~/.bashrc, and execute the commands in that file as if they were written directly into the script.
Here, source ~/.bashrc is telling the script to execute the commands in the ~/.bashrc file. This is typically done to set up environment variables, paths, and activate any software or tools that are required for the script to run successfully. In our case this tells Slurm where we have installed conda and thus enables Slurm to use conda itself.
This allows slurm to, after executing source ~/.bashrc, activates a Conda environment using mamba activate seqkit_2.6.1. This ensures that the SeqKit tool and its dependencies are available and properly configured for use in the subsequent part of the script.