mamba create --name snakemake -c conda-forge -c bioconda snakemake=7.32.4 python=3.11.6NanoITS
NanoITS is a classifier for long-read Oxford Nanopore data of the eukaryotic 18S/SSU-ITS1-ITS2 operon.
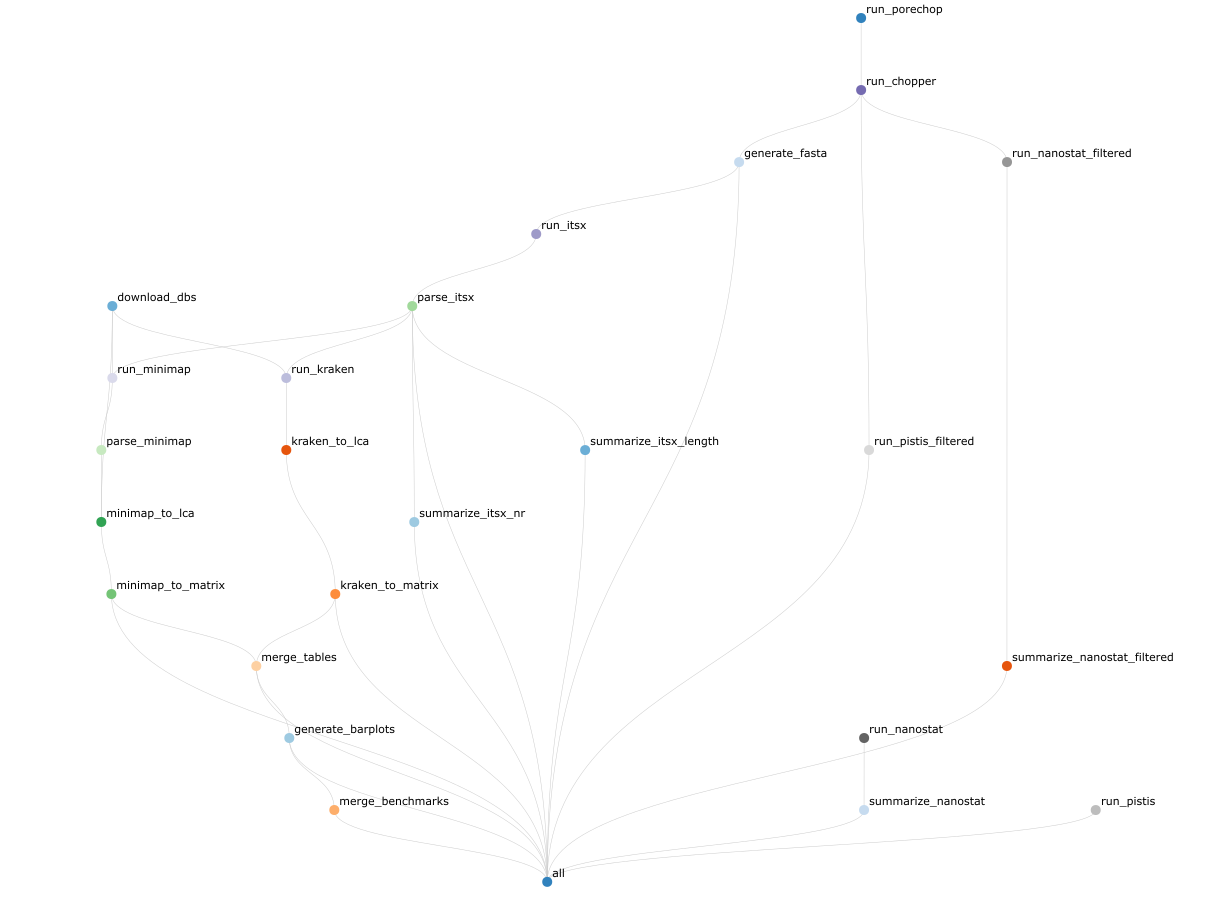
When giving the tool some nanopore long-read data it will:
- Provide a quality report of the raw reads
- Check the reads for adaptors and barcodes and if present trim the reads using porechop
- Remove low-quality and short reads with chopper
- Provide a quality report for the cleaned reads
- Identify and separate both the ITS1/ITS2 and 18S rRNA gene using ITSx
- Classify the SSU and/or ITS1/ITS2 gene using kraken2 and/or minimap2 and the Silva and Uniprot database, respectively
- Generate taxonomic barplots and OTU tables
Below you can find the full workflow:
Quick start
To run NanoITs, install conda and use it to installed snakemake as follows:
Afterwards, you can clone the directory from github via:
git clone https://github.com/ndombrowski/NanoITS.gitProvide your sample names and path to the samples as a comma-separated file. To view an example have a look at example_files/mapping.csv. The sample names should be unique and consist of letters, numbers and - only. The barcode column can be left empty as it is not yet implemented. The path should contain the path to your demultiplexed, compressed fastq file(s).
Adjust config/config.yaml to configure the location of your mapping file as well as specify the parameters used by NanoITs.
NanoITs can then be run with (adjust the cores based on your system):
#perform a dry-run to ensure everything is working correctly
snakemake --use-conda --cores <nr_cores> \
-s <path_to_NanoITS_install>/workflow/Snakefile \
--configfile config/config.yaml \
--conda-prefix <path_to_NanoITS_install>/workflow/.snakemake/conda \
-np
#run NanoITS
snakemake --use-conda --cores <nr_cores> \
-s <path_to_NanoITS_install>/workflow/Snakefile \
--configfile config/config.yaml \
--conda-prefix <path_to_NanoITS_install>/workflow/.snakemake/conda \
--rerun-incomplete --nolock